
How Nuria Lopez-Bigas is untangling the genetics of cancer
A scientist’s job, essentially, is to find patterns in data. But as we begin to collect more data than any one person can analyse alone, what if we let the patterns find themselves?
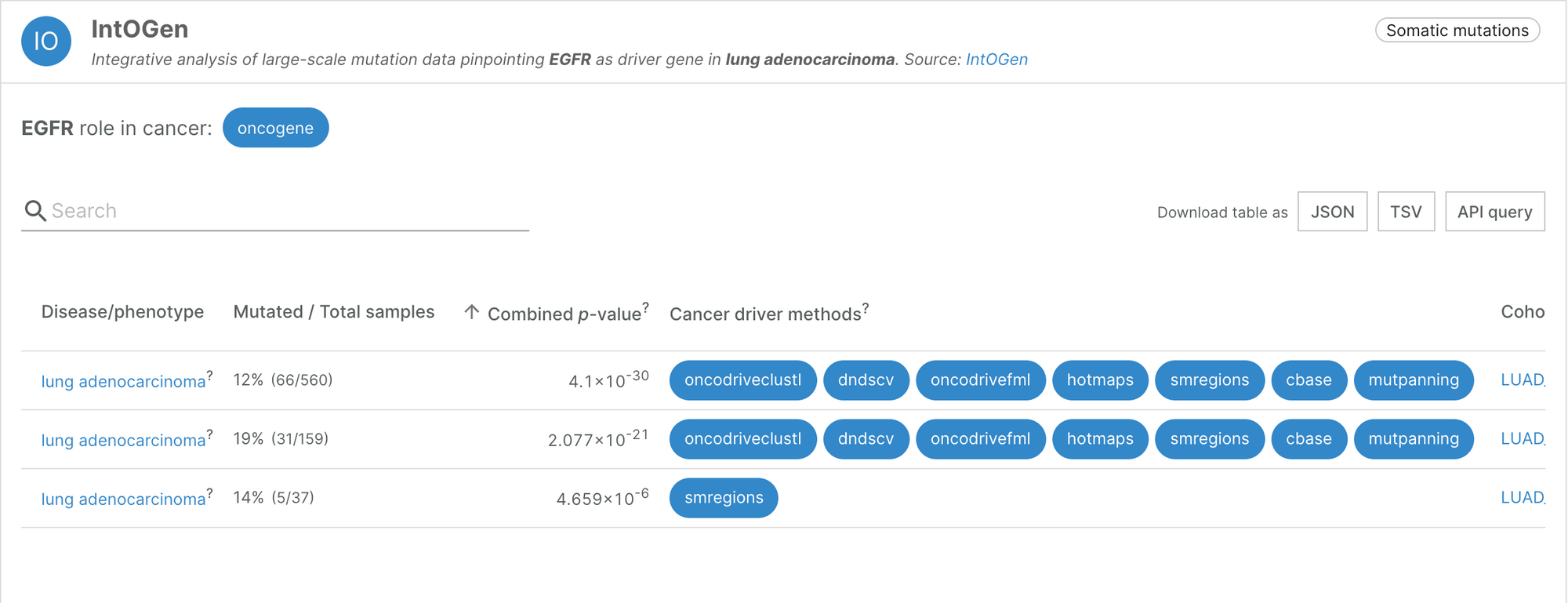
Núria López-Bigas is a molecular geneticist who transitioned to bioinformatics during her postdoctoral project at EMBL’s European Bioinformatics Institute (EMBL-EBI). She presented her work during the last Open Targets lab meeting of 2021. Her lab in Barcelona at the Institute for Research in Biomedicine studies cancer from a genomics perspective, focusing particularly on the identification of cancer driver mutations, genes, and pathways across tumour types with the aim of using this information for drug target discovery. Their framework to identify cancer genes, Integrative OncoGenomics (IntOGen), is one of the data sources of the Open Targets Platform.

Cancer is characterised by the abnormal and unregulated growth of cells in a tumour. Certain mutations in the DNA of tumour cells change the way that cell division is regulated, leading to runaway cell division. By understanding which mutations have occurred, the genes they affect, and how this alters the biology of the cell, we can begin to create targeted therapies. Several initiatives collect patient tumour samples and blood samples, so that cancer cell mutations can be identified by comparing the genome of cells in the tumour to the patient’s genome. These studies — a current total of 28,076 samples of 66 cancer types in 221 cohorts — represent a wealth of data that can be used to identify key DNA changes.
In fact, the challenge is not finding the mutations themselves, but finding the ones that drive a tumour cell’s cancerous behaviour. “Variation happens in all the cells in our tissues every day, due to a number of mutational processes,” explained Núria. Most of that variation — so-called ‘passenger mutations’ — has no effect. Of the thousands of differences between a patient’s tumour cells and other cells, only a handful of those differences are of interest to us. In total, 203,003,747 cancer cell mutations have been identified so far through these initiatives, and Núria’s challenge is to disentangle the driver mutations from the passenger ones. With her team, she decided to take an innovative approach: to learn directly from the mutations themselves.
The first challenge: identifying cancer driver genes
“The key was to think about cancer cells in terms of Darwinian evolution,” said Núria. Driver mutations will confer an advantage to the cell in which they occur, so within a tumour, they will appear in different patterns to passenger mutations. The first challenge Núria’s team faced was to identify, from the pattern of mutations within tumour cells, which genes had been strongly selected for. They created a method to find higher numbers or significant clusters of mutations, indicating the location of a cancer driver gene. The result of this analysis is a compendium of 568 cancer driver genes, available to explore within the IntOGen browser [1].
“One of the advantages of this approach is that we try to generate the most comprehensive and complete view, based on signals of positive selection, of which genes are involved in which tumour types,” explained Núria. This approach recovered almost all the well-known cancer driver genes, and identified some new ones. The systematic analysis also confirmed something that cancer researchers had long suspected: most of the cancer genes are very tissue-specific. Only a handful of genes are drivers of more than 20 tumour types; most of the genes identified drive the progression of only a few cancer types. There are also several genes that are frequently mutated only in very specific cancer types.
Additionally, one of the features stored by the IntOGen pipeline is the enrichment of mutations in particular domains for every protein and cancer type. These predictions reveal the most likely types of mutation to cause the cancer type, and whether it is conserved. This information is essential for drug design.
At least for the tumour types with more data, Núria believes that we have a good representation of the most frequently mutated cancer genes. The challenge is now to find, within those cancer genes, the specific mutations that are capable of driving the development of tumours.
 Francisco Martinez-Jimenez
Francisco Martinez-Jimenez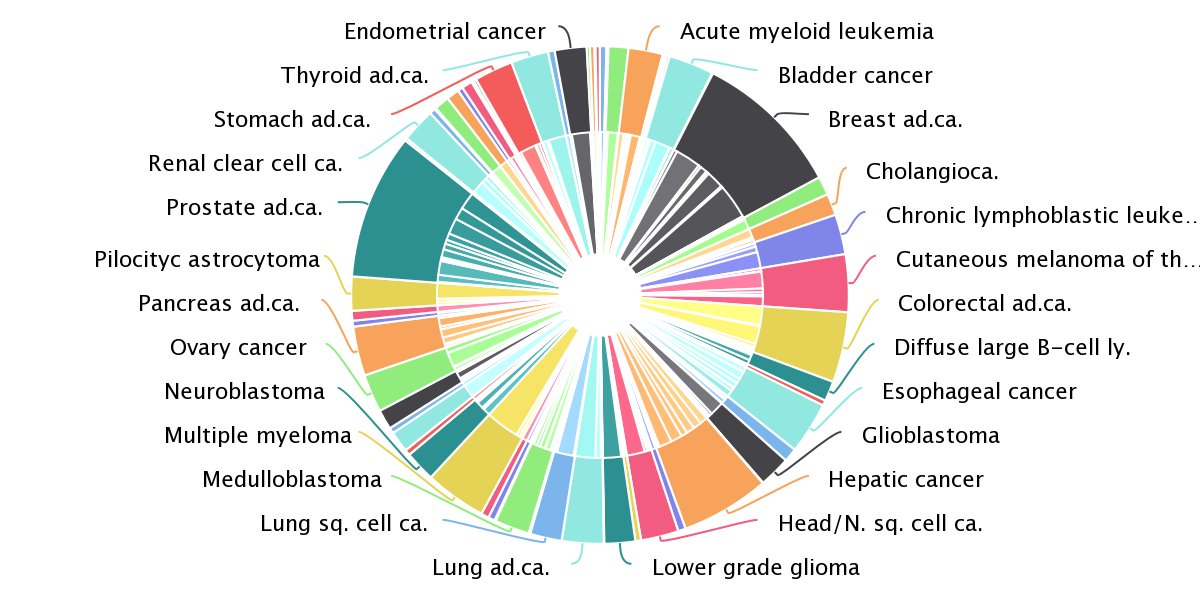
For a deep dive on IntOGen, check out our Q&A with Francisco Martínez
From cancer driver genes to cancer driver mutations
Here too, the team approached this in a systematic manner, learning from the data. “This is the best natural experiment,” said Núria. “All the tumours that have been sequenced give us information about this experiment that has occurred in the human population.” The team calls their in silico saturation mutagenesis project, BoostDM, “machine learning inspired by evolutionary biology.” BoostDM learns and stores the combinations of features that define driver mutations in each gene/tumour pair, such as the location of mutational clusters, the domains that have an enriched number of mutations, and the enriched mutation types for each of the genes. This information is all available to download. “We didn’t want this to be a black box, to have a score or a classification with no explanation. We wanted this to be something that you can understand.”
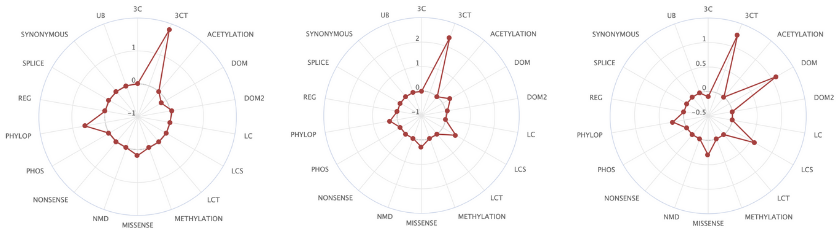
The 185 high-confidence gene/tumour type models created by BoostDM reveal some never-observed driver mutations, and show that for certain genes implicated in multiple cancers, the driver mutations differ depending on the type of cancer [2].
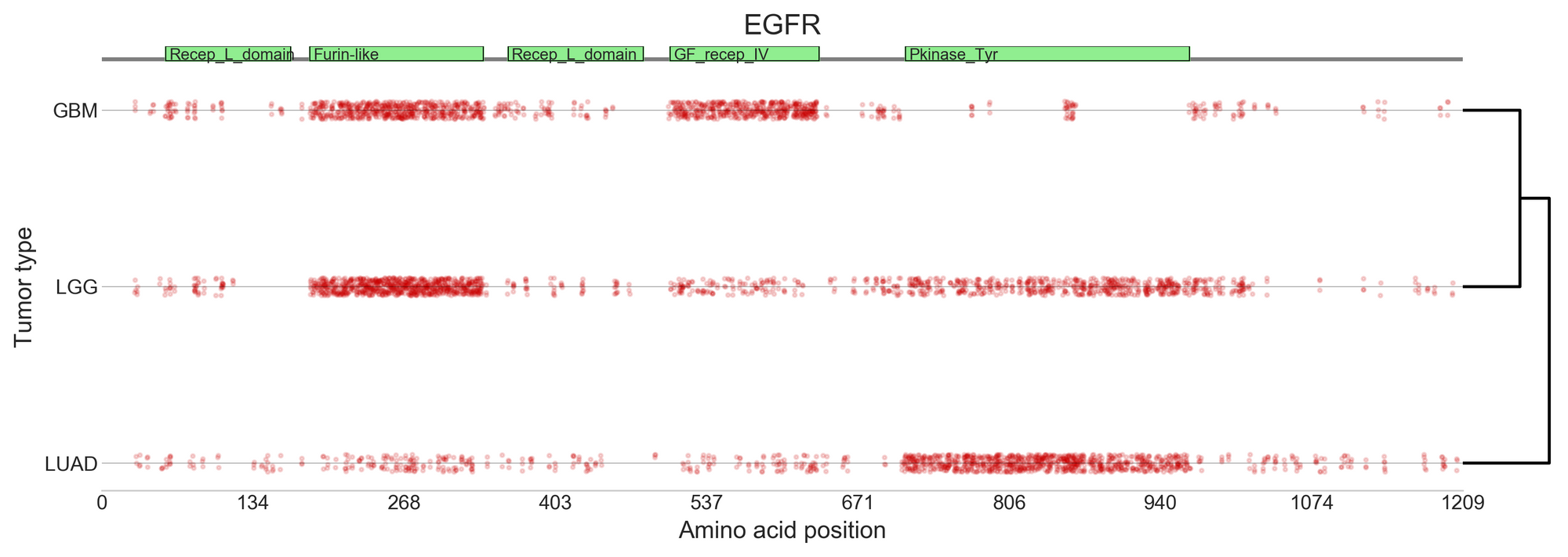
BoostDM predictions are further used in the Cancer Genome Interpreter, in combination with expert curated databases. From a list of mutations in a particular cancer type, the Cancer Genome Interpreter identifies which mutations are more likely to play a role in the growth of the tumour, and flags genomic biomarkers of drug response, to suggest anti-cancer therapies that could target those mutations in that context. This way BoostDM results help to interpret variants of uncertain significance, as an additional tool to help clinicians understand their patients’ data.
The blueprint maps of the locations of driver mutations within a gene are freely available for researchers to browse, and the information they contain has several implications for drug discovery. For example, this data could help identify a protein’s functional domain, with consequences for the design of compounds that could be used to target it. One example discussed in the round-table following Núria’s talk was Werner syndrome ATP-dependent helicase (WRN), identified as a target for tumours with microsatellite instability in an Open Targets project [3]. WRN has two enzymatic domains; the team disrupted each site to pinpoint the helicase domain as the protein’s functional domain. BoostDM could help resolve similar questions, identifying functional domains within a gene to suggest which might be the most important to target with a cancer therapy.
Future developments
“This is just the beginning, and there is a long way to go,” Núria emphasised, projecting an image from her home town of a path disappearing into rocky mountains. “For us, this is a proof-of-concept that the data holds a lot of information, and that we can go much further.” For example, there is still a lot to be learned from the context in which these cancers occurred. Ideally, the model would take into consideration the other mutations that have occurred in that cell, the stage of the cancer, and whether the patient has already received treatment. “In the future, we hope to be able to predict patient-specific mutations, to allow for more personalised medicine approaches.”

References
1. Martínez-Jiménez, F., Muiños, F., Sentís, I. et al. A compendium of mutational cancer driver genes. Nat Rev Cancer 20, 555–572 (2020).
2. Muiños, F., Martínez-Jiménez, F., Pich, O. et al. In silico saturation mutagenesis of cancer genes. Nature 596, 428–432 (2021).
3. Behan, F.M., Iorio, F., Picco, G. et al. Prioritization of cancer therapeutic targets using CRISPR–Cas9 screens. Nature 568, 511–516 (2019).


