Getting to know intOGen, a resource for the identification of cancer driver genes
A crucial goal in cancer biology is to identify genes with mutations capable of driving tumorigenesis.
In the last decade the advent of cancer genome sequencing has rendered an extraordinary opportunity for cancer researchers to identify new cancer driver genes by studying their unexpected pattern of somatic mutations across hundreds of tumor genomes. Driver discovery methods explore diverse positive selection signals, providing additional information about how genes are dysregulated in cancer. Therefore, the combination of complementary methods can bring the most comprehensive landscape of cancer driver genes and their mechanism of action in cancer.
In the Open Targets Platform, we've several datasets integrated in a single resource to allow for the identification of new genes in cancer (and other diseases), revealing new opportunities for therapeutic intervention in oncology. One of these datasets is intOGen.
I caught up with Francisco Martínez from the Biomedical Genomics Lab at IRB Barcelona to find a little more about intOGen and what its latest developments in identifying cancer driver genes are. I started our Q&A by asking:
What is the aim of intOGen and when was it first released?
IntOGen is an integrated resource for onco-genomics, whose aim is to collect and analyse somatic mutations from cancer genome sequences to identify cancer driver genes.
We first released IntOGen back in 2013, and since then we’ve been constantly adding new data to it. In recent years cancer genome sequencing data that is publicly available has increased at staggering pace.
At last but not least intOGen also allows to explore possible mechanisms of action across different tumour types.
Can you tell us some of the main challenges you face when retrieving and integrating data for intOGen?
I think the main challenge is time. We spend an astonishing amount of time gathering somatic mutations from varied sources of cancer genome sequences, and assessing whether the data we’ve downloaded fulfills the appropriate (and stringent) quality standards.
The good news is that we can count on large sequencing initiatives, such as the Cancer Genome Atlas (TCGA), the International Cancer Genomes Consortium (ICGC) and many others, to do the heavy lifting for us. These are well established, long-standing genomic resources with comprehensive molecular analyses on more than 10K patients, with samples from both tumour and their healthy tissue counterparts. Most of this data is available in a standard format, which makes our work more straightforward.
The latest version of intOGen (release 2020-02-01) contains more than 200 million somatic mutations and 568 cancer driver genes.
What are the specific steps when assessing the quality of these sequences?
Despite the availability of resources such as ICGC, TCGA and others, we still rely on separate sequencing projects, with differences in mutation calling methodologies, lack of standard formats or restrictive access rights due to publication agreements. Sequence data coming from these other sources needs to be preprocessed first, for example the genomic coordinates if on the GRCh37 assembly need to be liftover to GRCh38, before we can move on to identifying cancer driver genes.
So, the best call to action if you are generating new sequencing data from cancer genomes is to submit your data directly to platforms such as cBioPortal or NCI Genomics Data Commons. This will ease the access to the wider research community so that it can be used by other groups for downstream analysis, such as the one we perform with intOGen.
How does intOGen compare to the Cancer Gene Census from COSMIC?
The Cancer Gene Census is a resource that provides manually curated annotation of genes involved in cancer alongside cancer hallmarks. This information is curated from scientific literature. For more details, I’d encourage your readers to check your previous blog post, How to navigate the complexity of cancer genetics with the Cancer Gene Census.
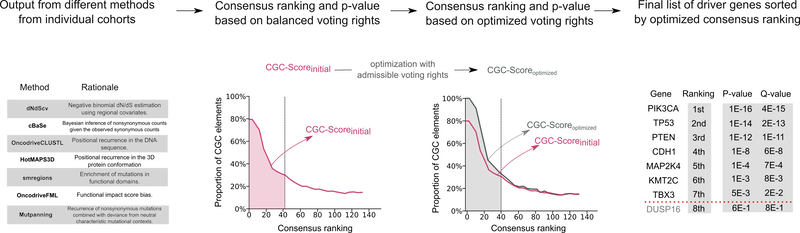
IntOGen, on the other hand, encompasses an automatic strategy that systematically identifies mutational cancer driver genes for each input cohort of patients by combining the output of multiple state-of-the-art methods.
It’s worth mentioning that we also take into account the data provided by the Cancer Gene Census. Firstly, for each cohort we analyse, we estimate the credibility of the intOGen methods based on the proportion of predicted cancer driver genes in our pipeline that are annotated in the Cancer Gene Census. We then combine the outputs of driver identification methods by weighting their predictions by its inferred credibility.
Combinatorial methodology applied with intOGen for the identification of cancer driver genes.
I believe that both resources complement each other quite nicely and can provide a comprehensive picture of the global landscape of cancer driver genes. It’s great the research community can now find both data sources integrated in one place, the Open Targets Platform.
Speaking about the Open Targets Platform, we are soon to release our latest update (release 20.02). Among other additions, we’ll incorporate the new intOGen data. What are the changes that our users should expect with this new data?
The latest release of intOGen includes a recently developed version of the pipeline that leverages seven state-of-the-art methods and a newly implemented combinatorial strategy. Check Which methods for cancer driver gene identification are used? to learn more.
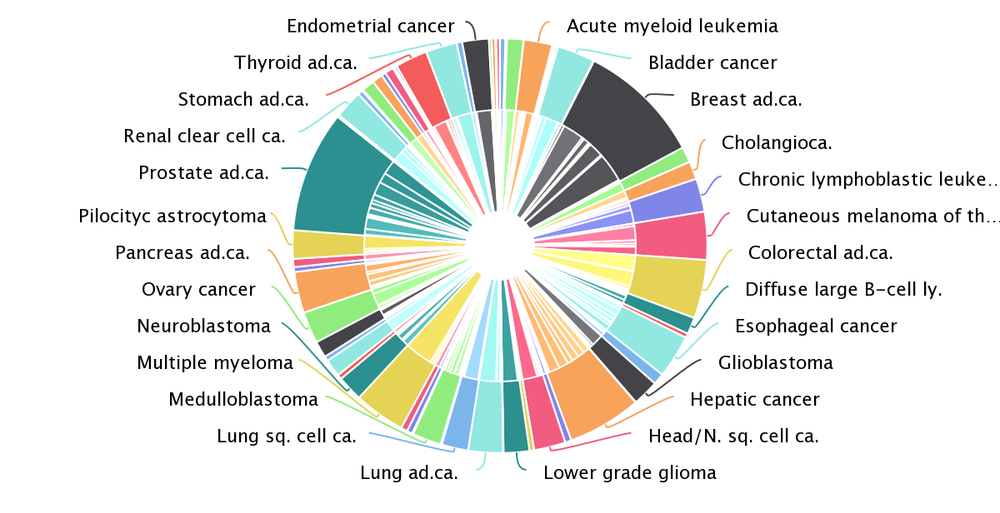
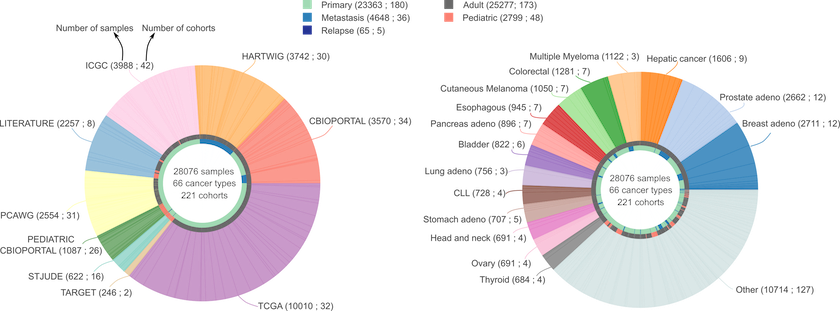
We have applied the new pipeline to identify cancer driver genes and their tumorigenic mode of action to more than 28,000 human tumor samples across 221 cohorts of 66 different tumor types, including the Pan-cancer analysis of whole genomes. We have paid extra care to include cohorts that tend to be underrepresented such as cohorts from rare tumor types, from pediatric patients and from metastatic tumors.
The new release of intOGen corresponds to the largest repository of automatically annotated cancer driver genes alongside their mutational features. We plan to periodically update the platform with new results as we get more samples from publicly available repositories. So watch this space!
I expect the new data will lead to new target-disease associations supported by intOGen somatic mutations, and it will likely increase the association score for associations previously described in the Open Targets Platform.
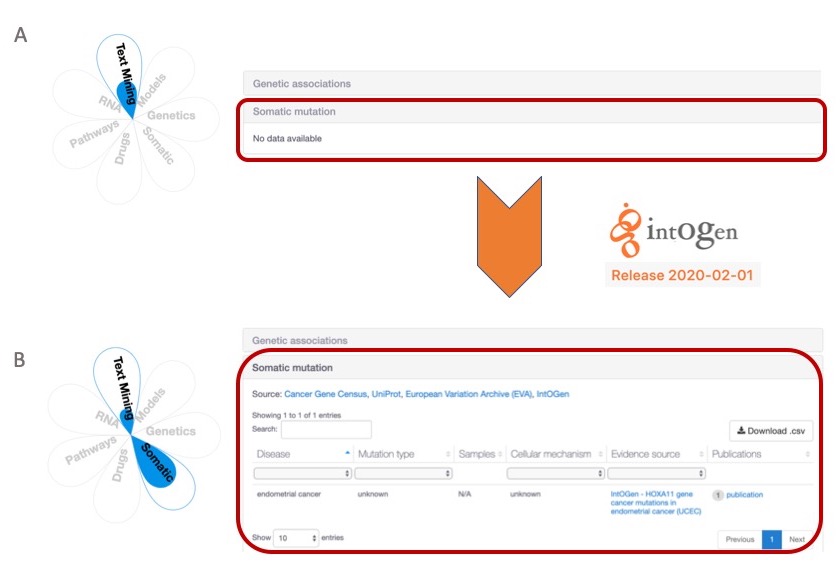
With the update to intOGen version 2020-02-01, we can now add extra support to target-disease associations. A) HOXA11-endometrial disease association is not supported by somatic mutations in the 19.11 release. B) The same target-disease association can now be supported by somatic mutations from the new intOGen data, which will be available in the Open Targets Platform release (20.02).
Looking forward to our new release then. Well, it's been great to learn more about intOGen. Thank you Fran for taking your time in this Q&A. Before we go, do you have any final comments?
Yes, I would like to stress that intOGen would not have been possible without the generosity of patients, clinicians and researchers that decide to share their samples for research purposes. We hope to make a convincing case that accessible data is necessary to enable and accelerate progress in science. Check our About page to stay tuned with upcoming publications.
Finally thank you, Denise and all the Open Targets team, for your fantastic work and for sharing intOGen results in your platform.
This blog is a joint contribution by Francisco Martínez and Denise Carvalho-Silva. You can contact Francisco and his colleagues at bbglab [at] irbbarcelona [dot] org.