How Ana Cvejic built an immune atlas of lung cancer
Lung cancer is the deadliest form of cancer worldwide. Non-small cell lung cancer (NSCLC) is the most common form, making up 85% of cases. In a paper published today in Nature Communications, Open Targets researchers and collaborators used single-cell RNA sequencing to create a high resolution molecular map of immune cells in NSCLC tumours, to better understand the role of these cells in disease progression.
Myeloid cells, frontline immune cells that initiate inflammation, are abundant within tumours. Recent research has shown that macrophages, in particular, can either suppress or promote tumour growth. Targeting these cells might be a potential therapeutic avenue for treating NSCLC, but there are many types of macrophages, and we’re only beginning to pick apart their different types and what pushes them towards one behaviour or another within a tumour.
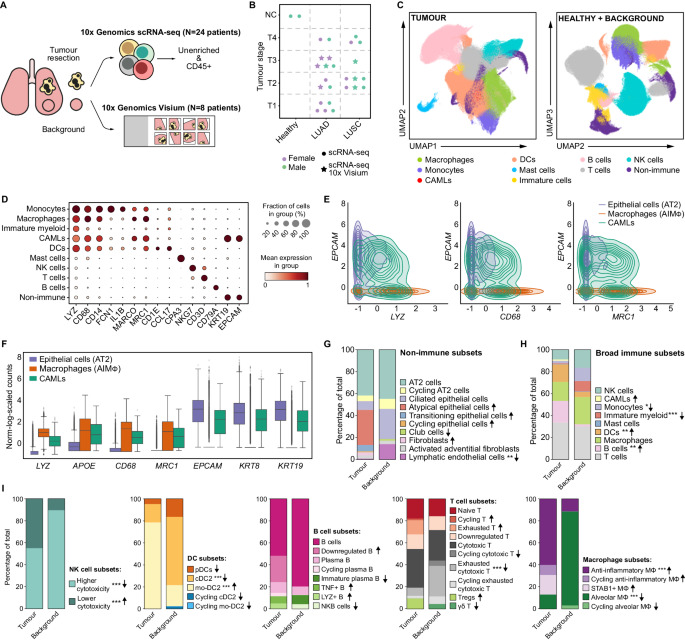
With nearly 900,000 cells from 25 individuals, this is the largest single-cell multiomics analysis of treatment-naive patients. Importantly, the samples include adjacent, non-tumour samples used as a control for the analysis.
A single-cell atlas of immune cells in lung cancer
“Our aim was to study the myeloid cells in the tumour environment, how they change and interact with tumour cells in disease,” explains Ana Cvejic, who led the project. “The more we know about what’s going on within tumours, and how this might differ from patient to patient, the better we can treat them.”

The team also performed spatial transcriptomics on a subset of the samples, to analyse the interactions between cells. Since the study was designed to capture as much information as possible, in doing so they created a comprehensive atlas of immune and non-immune compartments in lung cancer.
Additionally, NSCLC has often been studied as one disease, when in fact there are several subtypes. Of these, adenocarcinoma and squamous cell carcinoma are the most common, and from a molecular perspective, they are quite different diseases. Part of the aim of this study was to try to identify differences between these cancers at the single-cell RNA level, for example in terms of cell abundance, cell states, or ligand-receptor interactions. This could then pave the way for subtype-specific therapies.
What did they find?
“There were quite a few interesting findings,” Ana says. “When you have such a big data set and so many cell types, I think it's natural to get excited about various parts.”
Overall, the team were able to recapitulate recent findings showing an inverse relationship between anti-inflammatory macrophages and natural killer and T cells in the tumour. They also found many more types of myeloid cells in the tumour than in the adjacent tissue, and there were clear differences in the genes activated in myeloid cells, with a general shift towards cholesterol export in the tumour. This might play a role in supporting the growth of the tumour cells, since cholesterol is key to cell division.
“One of the most interesting findings is that some of the cells had a foetal-like phenotype, which supports some very recent findings in the field that myeloid cells in a tumour environment can be reprogrammed to a foetal state,” Ana explains.
Ana’s team were also able — for the first time — to fully transcriptionally characterise cancer-associated macrophage-like cells (CAMLs) found within the tumour samples. These unique cells have been observed in blood samples of patients with various malignancies, including NSCLC. They are interesting because they have characteristics of both myeloid (immune) cells and epithelial (lining) cells.
“This type of cell has only recently been described, so we were very excited, but we also had to be very cautious to make sure that we were seeing something real. It was a mixture of frustration and excitement, and we spent a significant amount of time reassuring ourselves that these are real, at least on a computational level,” Ana says.
Finally, the researchers suggest potential new subtype-specific therapeutic targets. Although there weren’t significant differences in the cell composition between adenocarcinomas and squamous cell carcinomas, they observed differences in the interactions between tumour cells and the surrounding immune cells between the two subtypes. In particular, they identified various immune checkpoint inhibitors, including some potentially novel ones which they suggest could be an interesting therapeutic avenue.
The design of the study was deliberately comprehensive, so there is lots more information to dig out from this dataset. And in fact, that’s exactly what I’m hoping will happen. — Ana Cvejic
A collaboration with industry scientists
As part of Open Targets, Ana’s team worked closely with pharmaceutical partners, particularly Sanofi and GSK. “The Open Targets partnership is a fantastic opportunity to create and collaborate on innovative projects such as this one, which we wouldn’t have been able to do alone,” says Angela Hadjipanayis, Head of Disease Profiling Genomics & Flow Cytometry Precision Medicine and Computational Biology led by Emanuele di Rinaldis at Sanofi.
“We’re very interested in reference datasets like these that create large amounts of multimodal data on a given indication, where we can discover new mechanisms to target, hopefully leading to new therapies.”
“This atlas provides key insights into the tumour microenvironment in non-small cell lung cancers, from which we can back-translate clinical trials to better understand our results. It’s an excellent foundation to inform our drug discovery initiatives and dig into the biology of potential new targets.”
Applications to future research
The data is now publicly available for the broader scientific community to explore and follow up. “You could spend years analysing this data! The design of the study was deliberately comprehensive, so there is lots more information to dig out from this dataset. And in fact, that’s exactly what I’m hoping will happen,” Ana says.
“Beyond that, I think sequencing is a very powerful approach to get to the bottom of what’s happening in disease, especially if you can include more modalities. Here we used single-cell RNA sequencing, but you could add another layer of understanding by, for example, using chromatin accessibility studies to find out what’s happening at the epigenome level.”
“And ultimately, a lot of this is down to numbers. Although this is the largest dataset of its kind, we would probably benefit from an even bigger dataset. These are not inbred mice, these are individuals with different backgrounds and histories, so the more people you can include in your study, the better you can pick up genuine differences between disease and control tissues.”
“And now that researchers are starting to use AI to learn from data, it’s important that the datasets are sufficiently big and the experimental design is made in such a way to permit that. Of course, sequencing is what my lab does, so I’m a little biassed!”