Getting a finer view on genes leading to Alzheimer’s disease risk
“Developing treatments for Alzheimer’s disease is notoriously difficult,” explains Jeremy Schwartzentruber, a statistical geneticist at Open Targets.
One reason for this is that Alzheimer’s is a late-onset disease, so it is complicated to accurately represent the mechanisms of the disease in the lab, and research is less likely to translate to effective therapies. However, disentangling the genetics of the disease can identify genes that are likely to be causally involved in the pathology, hopefully pointing the way to effective therapies.
By comparing large numbers of people with the disease to those without, genome-wide association studies (GWAS) identify genetic regions with differences between people that lead to risk or protection from the disease. Most genetic differences identified this way do not directly alter genes that encode proteins, but affect non-protein-coding regions. The major challenge for GWAS studies is therefore to identify which specific gene in the region is influencing disease risk. So far, only a few genes have been identified that clearly influence the risk of developing Alzheimer’s disease.
In a paper recently published in Nature Genetics, Schwartzentruber and his colleagues found 37 regions associated with Alzheimer’s disease, including 4 regions which hadn’t previously been identified. Their research produced a prioritised list of genes within those 37 regions that may potentially alter Alzheimer’s disease risk.
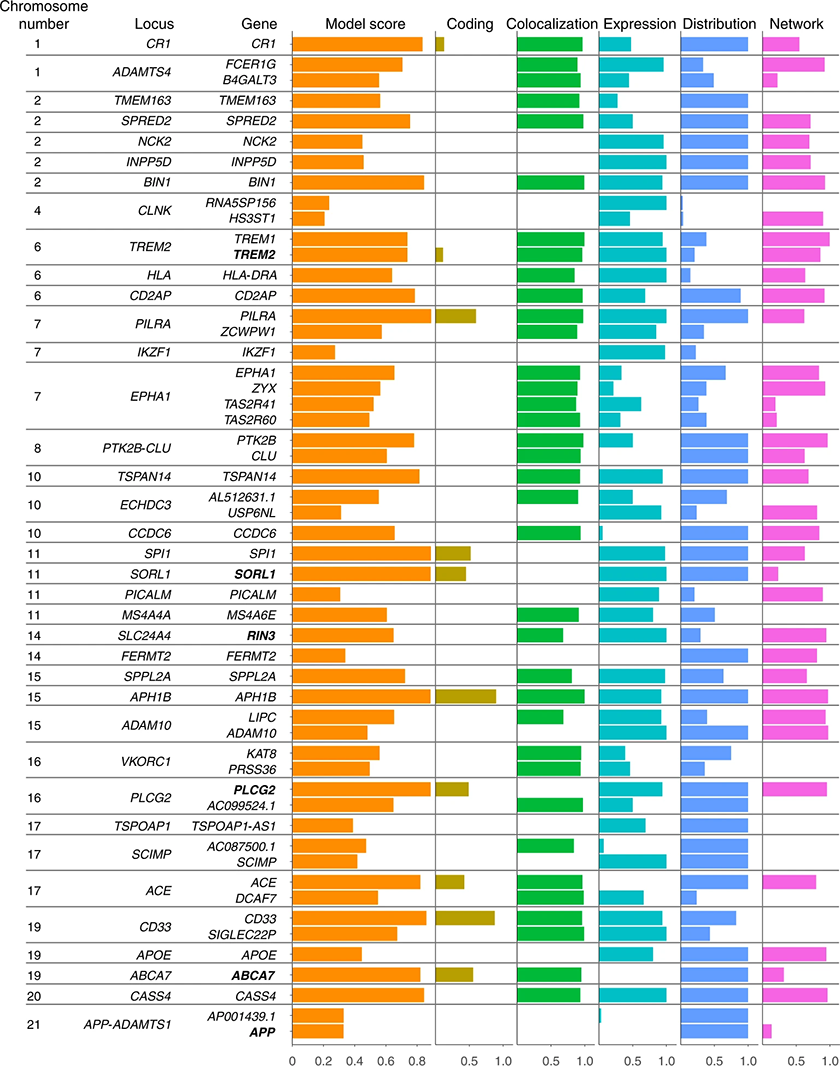
A selection of the top prioritised genes at genome-wide significant Alzheimer's disease loci, and the score components for each gene
“What is most unique about our study is that we integrated multiple layers of ‘-omics’ data to prioritise the causative genes at each of our GWAS regions,” says Schwartzentruber. By implementing a comprehensive and rigorous analysis, the team aimed to generate a quantitative score for each gene in order to rank the most promising ones for follow up studies. They also took advantage of a new study design which uses data from both Alzheimer’s patients and from larger studies of people without dementia, but who report first-degree relatives as having Alzheimer’s disease. First-degree relatives share 50% of our DNA, and can thus be considered proxies of the disease. Using this new design enabled the team to considerably expand the pool of available data.
“We found that it’s important to integrate different types of data to get the best prioritisation of genes possible,” explains Schwartzentruber. The first of these types of data comes from genetic colocalization tests, which determine whether the risk for Alzheimer’s disease in a particular region of the genome has a shared genetic basis with control of gene expression in different tissues (known as Quantitative Trait Locus studies). They then added another three layers of data.
The second type of data came from a technique known as ‘fine-mapping’: the team assigned a probability to each genetic variant in a region for being the one driving the association. In some cases fine-mapped variants lay directly within a good candidate gene. The third type of data came from gene networks based on protein-protein interactions. This can tell you whether one gene in a region has strong interactions with other known or suspected Alzheimer’s disease risk genes. The final type of data was single-cell gene expression from human brain cell types in the Allen Brain Atlas. Integrating this information allowed the team to determine that the only cell type clearly enriched for Alzheimer’s disease risk was microglia, the native immune cells of the brain.
One of the most intriguing associations they describe was for TSPAN14, a gene which interacts with another newly-identified Alzheimer’s disease gene, ADAM10. “Our fine-mapping resolved to a few potentially causal variants, but deeper investigation showed that one of these (rs1870138-G) alters an invariant position of a binding motif for the transcription factor TAL1,” commented Schwartzentruber. “Since TAL1 is highly expressed in microglia, this could be a mechanism that affects TSPAN14 expression in microglia, and hence Alzheimer’s disease risk.”
This prioritised list of genes associated with Alzheimer’s disease will now need to be followed up to identify their potential mechanisms of action. Schwartzentruber and his colleagues are passing the baton down the translational pipeline: new experimental projects within Open Targets will track disease-associated phenotypes in cellular model systems, such as by knocking out specific Alzheimer’s disease risk genes and observing the effect this has on the ability of microglia to phagocytose substrates, or on the vulnerability of neurons to stressors.
“I think that there is still a ways to go in identifying all the Alzheimer’s disease risk genes, and continuing to expand the scale of case-control studies is worthwhile, as others have done recently,” says Schwartzentruber. “We can also do better at identifying causal variants at each disease-associated region, by using neural network models that predict the effects of every genetic variant or transcription factor binding. Many groups are continuing to produce better single-cell datasets of gene expression, chromatin accessibility, and other -omics that will help to get more confidence in disease mechanisms at specific genomic regions.”
More experimental research at Open Targets