Predicting Chemical Safety for Drug Discovery
The ability to forecast the success of new therapeutic drug is crucial to minimise attrition rates and to reduce costs and time spent in pharma R&D. How can we tap into the diversity of biomedical big data to gain useful insights in the different stages of drug discovery? Harnessing advanced computational resources and applying artificial intelligence (AI), especially machine learning and deep learning, in several stages of drug discovery is one approach that is gaining increased attention.
If you think that AI and ML are new fields, think again! They have been applied for decades in different domains, from playing chess to natural language processing. However, we have recently seen a lot of use cases in biomedicine and translational research. This is mainly due to advances in hardware, which enables new software approaches (e.g., deep learning), and availability of large volume of biomedical data amenable for training machine learning models.
I have recently joined Open Targets after completing my PhD at King Abdullah University of Science and Technology (KAUST), and in this blog post, I will highlight my previous work using ML in drug safety assessment during preclinical stages.
Drug Toxicity
We can define toxicity is a measure of any undesirable or adverse effects of chemical compounds. Specific types of these adverse effects are called toxicity endpoints, such as carcinogenicity or genotoxicity, and can be quantitative (e.g., LD50: lethal dose to 50% of tested individuals) or qualitative, including binary (e.g., toxic or non‐toxic) or ordinal (e.g., low, moderate, or high toxicity) endpoints.
Toxicity testing is a crucial step in drug discovery. Unfortunately, toxicity testing does not come cheap when the global in vitro testing market expected to reach $8 billion by 2023. Can we strike the right balance between the importance and costs of drug toxicity? How can we reduce the costs of toxicity testing and at the same time provide more accurate predictions to tackle attrition in clinical trials? One way we can address these challenges is to apply computational methods which will
- Improve toxicity prediction and safety assessment
- Reduce the need for animal testing
- Guide toxicity tests to prioritise chemicals for drug development

Predicting Drug Toxicity
Mathematical models have been applied for assessing compound toxicity since as early as 1935. Since then, we have seen a dramatic improvement of computational tools for gathering, modeling, simulating, and visualising toxicity data. We now have several methods to generate toxicity prediction models at our disposal.
Current in silico toxicology methods (e.g., QSAR, read across, and dose-response models) use compound feature vectors to predict compund toxicity. These methods aim at developing models to predict only one single toxicity endpoint, so we need to generate one model for each endpoint. This is a laborious and computationally intensive process when the number of toxicity endpoints is large. Additionally, the single toxicity endpoint approach lacks the ability to take into account possible correlations between toxicity endpoints. There is strong evidence of correlations between toxicity endpoints as demonstrated by FDA studies on rodents. For example, gene mutation, DNA damage, clastogenicity, and reproductive toxicity endpoints correlate with carcinogenicity.
We are now interested in generating models to predict several toxicity endpoints by utilising correlations between them. Multilabel classification methods generate models that learn from compounds features as well as correlations between toxicity endpoint. This can improve performance and reduce the number of generated models, making multi‐label classification methods more efficient and practical.
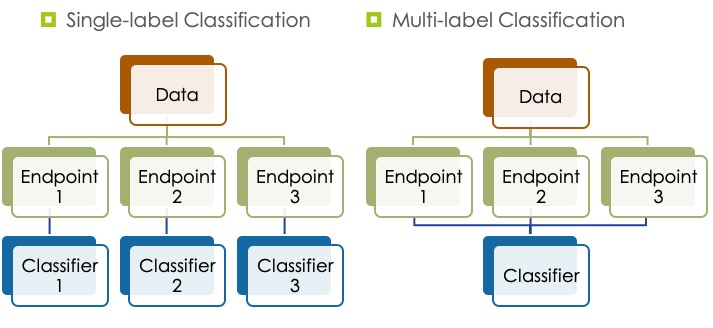
Developing multi-label classification models has its own challenges though due to missing labels in toxicity data sets. We may have unknown or missing toxicity profiles across all toxicity phenotypes, either because such data is unavailable (e.g., compounds are not tested for all toxicity phenotypes), or it may be hard to find (e.g., toxicity data is scattered in scientific literature). For example, the figure below shows that the first compound is not carcinogenic, but it causes developmental and maternal toxicity, but it is unknown if the compound is genotoxic or not. If the toxicity data is not available for all toxicity endpoints, we cannot easily determining the endpoint relationships.

Machine Learning Models for Predicting Chemical Toxicity
I performed comprehensive benchmarking and analysis of 19,186 multi‐label classification models I generated using various combinations of computational methods. I created a dataset that consists of toxicity profiles of 6,644 compounds in 17 toxicity endpoints in five species, compiled from several public toxicity databases. I used the SMILES notation for generating the compound's features. I generated categorical and structural features that may provide insight into mechanisms of action, including but not limited to:
- electrophilic, nucleophilic, and covalent reactivity mechanisms (e.g., Michael acceptors);
- radical mechanism by radical oxygen species formation;
- the compound's potential to bind/interact with biological entities (e.g., DNA, proteins, peptides, or estrogen receptors);
- bioavailabity, biodegradation, bioaccumulation, and stability;
- functional groups (e.g., hydrocarbons; halogen; bases and acids groups; and oxygen, nitrogen, sulfur, phosphorus, or boron groups); and
- classes of compounds.
The figure below shows accuracy scores per endpoint of the top‐ranked models generated by each multi‐label classification method in (a) internal validation and (b) external validation using an independent testing set. Rows correspond to the multi‐label classification methods whereas columns correspond to the 17 toxicity endpoints. Each cell shows the accuracy scores of each method per endpoint. The scores range from 0.0 (worst performance) to 1.0 (best performance). The best model I generated using the Random K Labelset algorithm achieved accuracy score of 90% in predicting the 17 toxicity endpoints. Check my publication, for a detailed description of the methods and analysis.
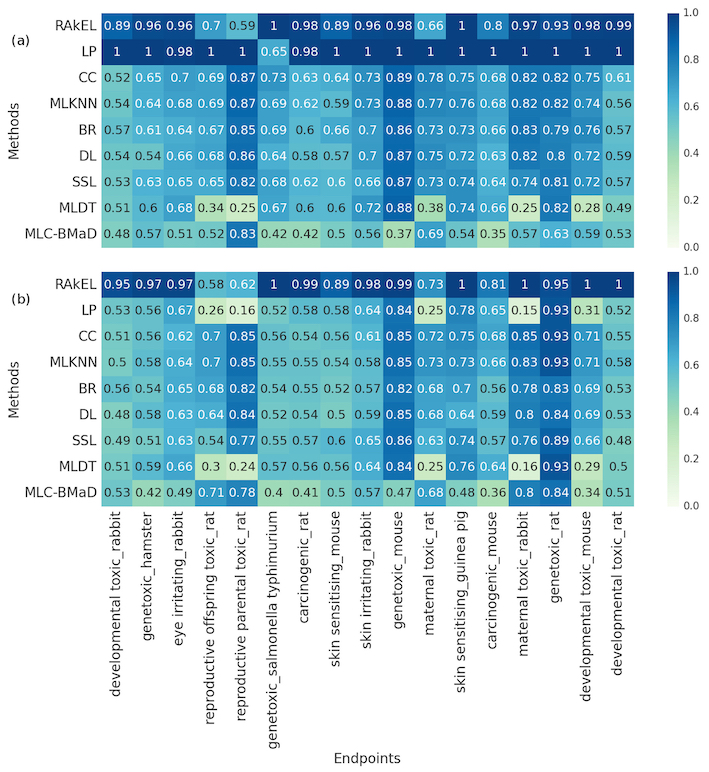
Part (a) The performance using cross-validation. Part (b) the performance using a hold-out testing set. (Abbreviations: BR, binary relevance; CC, classifier chains; DL, deep learning; LP, label powerset; MLC‐BMaD, multi‐label Boolean matrix decomposition; MLDT, multi‐label decision tree; MLKNN, multi‐label K nearest neighbor; RAkEL, random K labelset; SSL, semi‐supervised learning)
With this work, I have illustrated the advantages of using multi‐label models for compounds toxicity assessment even when toxicity data is partially available. The results of this comprehensive analysis of the state‐of‐the‐art methods for multi‐label classification indicates that combining endpoints correlations with compound features can increase model performance.
This is just the beginning! We can apply these predictive toxicity methods for target discovery. Just like a drug is associated with several toxicity endpoints, a target is associated with several diseases. We have already started exploring this space with our proof-of-concept LINK, the Open Targets Literature Knowledge Graph. Stay tuned for the upcoming developments on this theme and application to the Open Targets Platform.


