Open Targets Platform: adding FDA data with Scala and Spark
Open Targets Platform is an open-source project that brings together diverse biomedical databases to facilitate researchers in their search for new and effective targets for drugs. One of the challenges facing the core team of Open Targets is integrating and processing the large amount of publicly available biomedical data so that it can be used by stakeholders in their research. This blog post gives a short update on how the Open Targets core team integrated the United States' Food and Drug Administration's (FDA) Adverse Event Report System (FAERS) into the Open Targets data ecosystem.
What is Open FDA?
Since 2014 the FDA has operated the Open FDA project with the goal of increasing access to public data and contributing to public health outcomes. FAERS is a database of adverse event and medication error reports submitted to the FDA; for example when someone suffers nausea or some other adverse event while prescribed a medication. Medical professionals and consumers can voluntarily file reports of adverse medical events which are suspected to be associated with a drug. As a voluntary reporting scheme to which anyone can contribute the raw data needs to be treated with care. The FDA includes a number of disclaimers regarding the data including that the existence of a report does not establish causation, and that the information in the reports has not been verified.
The publicly available API provides nearly 12 million records of adverse drug reactions, approximately 130GB of data. To give some sense of the scale, if a person were to read one record per minute it would take over 20 years to consume all of the information.
Converting the raw data into insightful data
The core team prepared a pipeline using Apache Spark and Scala making it possible for us to analyse and extract insights from this information in minutes using Google Cloud's Dataproc service.
To clean the data, we limited the results to reports which did not result in patient death, and that were reported by a medical professional. This reduced the data set by approximately half. We further ignored specific events which were not related to the action of the drug, but rather to treatment, technology or human actions. The full list of excluded events can be found in the blacklisted_events file in the project's repository. After these steps, we were left with approximately 55 thousand unique drug-reaction pairs including 4,701 unique adverse events, covering 465 biological compounds.
Drugs which are often prescribed are going to appear more often in the database than seldom used ones, and show a greater variety of adverse effects. To take this into account we implemented a likelihood ratio test as proposed in "A likelihood-based method for signal detection with application to FDA's drug safety data". This methodology uses Monte Carlo sampling to test the relevance of each adverse effect associated with a drug, taking into account how often the event and the drug appear in the data set.
If the observations in the FAERS data exceed a specified configurable threshold (p = 0.95), we can say that the adverse event reports indicate a significant interaction with a drug. After filtering out non-significant results, we are left with a useful guide as to which adverse events are strongly associated with specific drugs. By linking the reported adverse events with medical compounds, researchers might be able to identify new linkages between specific targets and drugs and their effects.
One example is the case of Rofecoxib which was withdrawn from the market due to cardiotoxicity events. We can see in the results from the pipeline that this drug has multiple significant adverse events well above the critical threshold.
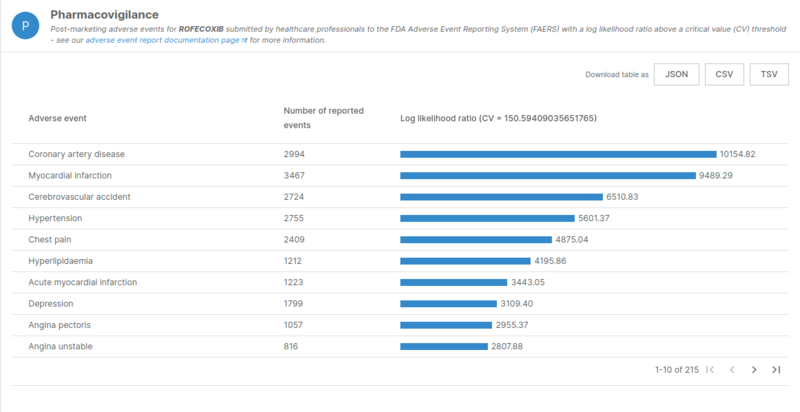
Log-likelihool ratios of adverse events related to Rofecoxib
Some thoughts on technology
It is worth taking a moment to consider the benefits Spark brings to our work. Spark is a cluster-computing framework that allows us to interact with our data through a programming interface. In many cases, this abstracts away the need to be concerned with low-level primitives related to memory management, IO operations and distributed systems among others. Within a few dozen lines of Scala code running on the Spark framework, we were able to prune 130GB of raw data into less than 10MB of key insights which drug discovery scientists can use for their research or be presented on the Platform’s website.
As an example of the utility of this framework, consider that the raw FDA source files are broken into 900 files. With the following few lines we can read the entire data-set into a Spark DataFrame, selecting only the columns we want without worrying about memory management:
val fda = ss.read.json(path)
val columns = Seq("safetyreportid",
"serious",
"seriousnessdeath",
"receivedate",
"primarysource.qualification as qualification",
"patient")
fda.selectExpr(columns: _*)
Behind the scenes, Spark will distribute this data making full use of the computing power available. Within the program, we can refer to the value fda as if it were a standard Scala collection, even though it is actually distributed across an arbitrary number of computers. Spark lets us focus on manipulating our data, rather than wrangling our resources.
Where there are not in-build library functions available, Spark offers user-defined functions which allow us in many cases to integrate our regular Scala (and Java) libraries into the Spark pipeline!
Releasing the package as a Fat-JAR with Spark embedded makes it possible to either run the entire pipeline on a single machine (without having to install Apache Spark) or on a cluster of processors (deploying the same package on a fully-managed Hadoop cluster becomes a configuration issue) to enable rapid completion of tasks. This gives us the flexibility to run the program on the computing resources available without having to rewrite any of the code itself. It also means that our users can run the pipeline themselves with their own parameters to meet their research goals.
The graph below shows how the time to complete this task varies using horizontal and vertical scalings: cluster template C1 shows the impact on elapsed time using horizontal scaling (adding more nodes to the cluster: more computers), and C2 shows the same effect when we scale the cluster vertically (reconfiguring the node with more cores: bigger computers). In both cases, we see decreases in analysis time with the application of additional computational resources. We eventually reach a hard limit where horizontal scaling stops seeing improvements as network and cluster management delays remove any benefit from additional resources.
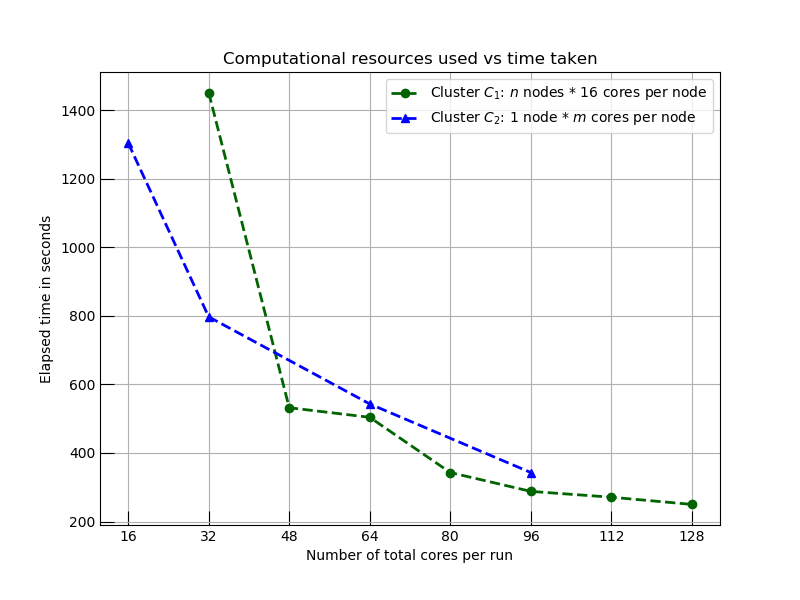
Comparison of two computing models on execution time
Running your own analyses
If you would like to explore the source code it is available from the Open Targets’ GitHub page. The jar file available in the releases section includes all dependencies needed to run the pipeline locally (without installing Scala or Spark) on the Java Virtual Machine; you just need to complete a configuration file specifying where to find the inputs and where to save the outputs. The significance thresholds and granularity of the sampling are also configurable to allow researchers to generate the data they need for their research with a minimum of effort.
The jar file can be run with the following command (as you can see from the README file):
java -server -Xms1G -Xmx6G -Xss1M -XX:+CMSClassUnloadingEnabled \
-Dlogback.configurationFile=application.xml \
-Dconfig.file=./application.conf \
-classpath . \
-jar io-opentargets-etl-backend-assembly-openfda-0.1.0.jar
Where the application.conf file is used to specify inputs, outputs and the parameters for the Monte Carlo sampling. An example application.conf file is provided in the readme.
Next steps
As part of our Open Source commitment, all code has been released to the community allowing for further development of new analytical strategies on adverse event reports. The combination of scalable technologies and cloud computing will constitute one of the pillars of our technical development, in order to bring large scale data-sets and high throughput computation to the widest community. While the Open Targets Platform team continues to identify and integrate new data-sets, we are moving more of our pipeline into Spark jobs which can be executed independently and are easy to scale both vertically and horizontally. Independent, flexible execution allows our users to focus on the parts of the pipeline that are of relevance to their research, and allows us to update data with greater regularity: when new data becomes available we can run the relevant pipeline steps without having to regenerate all of our analysis.
The backend team is currently working on migrating the API to the Play Framework and exposing endpoints using GraphQL, while the front-end team is busy making sure that all our users can explore and extract meaning from the data made available through the Platform.
These changes to our technical infrastructure will facilitate the rapid integration and updating data-sets making it easier for users to access that data through the Platform or our API. And with new data, users can uncover more novel target-disease associations and develop new therapeutic hypotheses that can be used for further downstream analysis and integration into drug discovery pipelines.
And if this sounds like something you want to be a part of - helping scientists with tools that enable them to browse data and generate and answer research questions - then head to our Jobs page or contact us.
We’re always on the lookout for talented software developers, bioinformaticians, and researchers that want to join our dynamic, international, and interdisciplinary team based on the Wellcome Genome Campus.