In-GenIE-ous experiments to find causal variants from GWAS
Genome-wide association studies (GWAS) have discovered thousands of regions of the genome associated with human disease risk. But each association typically reveals anywhere from a handful to many dozens of genetic variants that are all correlated with disease risk. In general only one or two of these variants are likely to actually cause the difference in risk between people, yet it’s notoriously difficult to find the causal variant(s). Most associated variants, whether single-nucleotide polymorphisms (SNPs) or short insertions / deletions, are in non-coding regions, and so they must somehow alter gene regulation rather than protein structure.
We developed a new method that can be used to screen up to a few dozen SNPs for a causal effect on gene transcription. We call it GenIE — Genome engineering-based Interrogation of Enhancers, and it was recently published in Nucleic Acids Research.
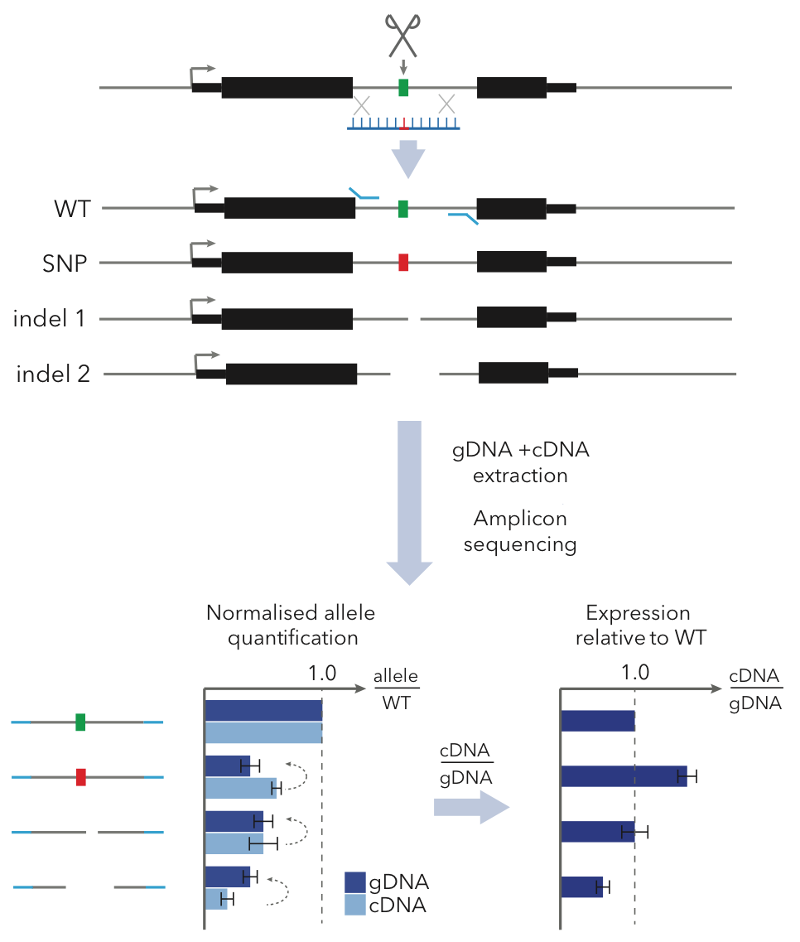
With GenIE we use CRISPR-Cas9 to target a double-stranded DNA break near the SNP of interest, and use homology directed repair (HDR) with a designed oligonucleotide to introduce alternative SNP alleles. We then extract genomic DNA and RNA (to make cDNA) from the edited pool of cells and do a simple PCR across the SNP of interest, followed by sequencing. Comparing the ratio of SNP alleles in cDNA vs. gDNA tells you the relative transcription levels of the two alleles — in other words, the causal effect of the SNP on gene expression.
We applied our GenIE method to functionally test likely causal eQTLs in iPSCs. We tested a SNP in MUL1 and one in ABHD4 and in both cases saw a strong effect of the SNP in the same direction as the eQTL.
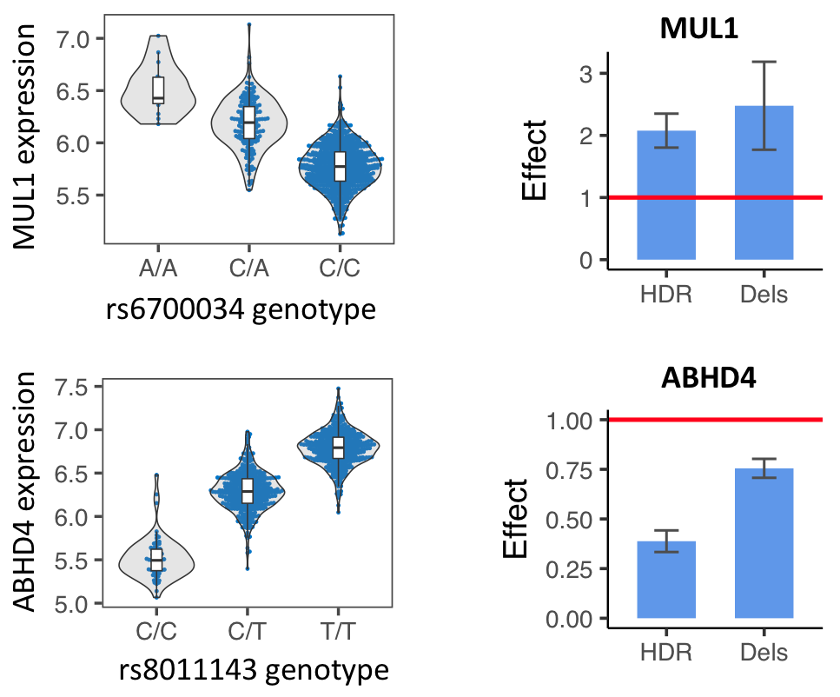
Left: eQTLs for MUL1 or ABHD4, showing expression of the different alleles.
Right: GenIE results, showing the relative expression of the HDR-introduced alleles (A vs. C for MUL1, and C vs. T for ABHD4) or the deletion alleles, relative to the reference allele.
Because we use Cas9, we are able to analyse both HDR events (with the desired allele) as well as deletions of different sizes covering the Cas9 cut site. In our MUL1 and ABHD4 examples, you can see that the deletions over this region of the genome also have an effect on the expression of the gene, which suggests that this region acts as an intronic enhancer.
You can use our R package, rgenie (available on CRAN), to plot the deletion alleles in your cDNA or gDNA.
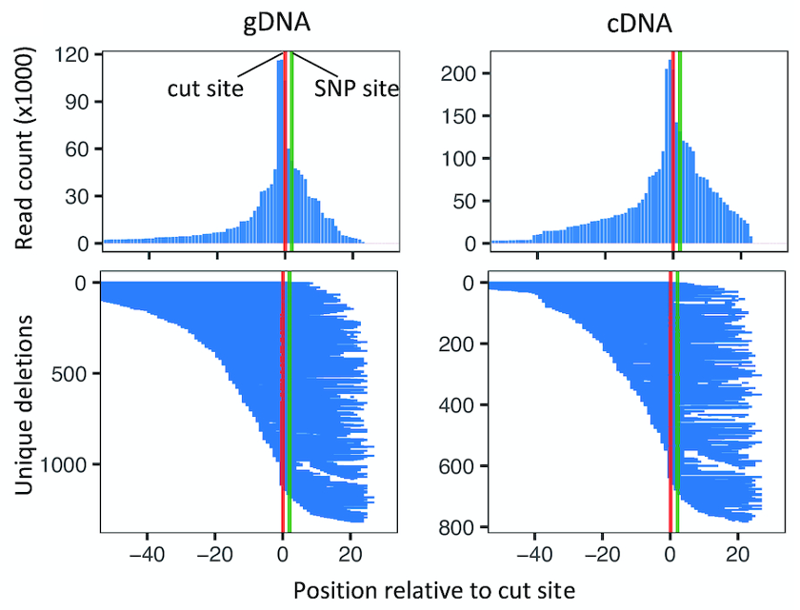
To see if deletions are having an effect, it helps to also compare the deletion profiles of individual replicates. Here is an intronic SNP in the CLU gene where there is a clear effect of deletions increasing RNA transcription by about 1.3x (cDNA replicates in red, gDNA replicates in blue).
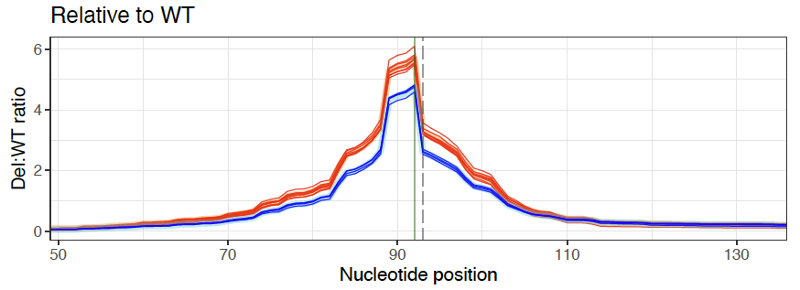
GenIE is especially sensitive to splicing changes if you position the PCR primers so that one is in the alternatively spliced exon. For example, we targeted a synonymous SNP in CCDC6. We didn’t see any effect of that SNP on splicing or transcription, but deletions that spanned the nearby splice site had a huge effect on transcription across the splice site, as you would expect.
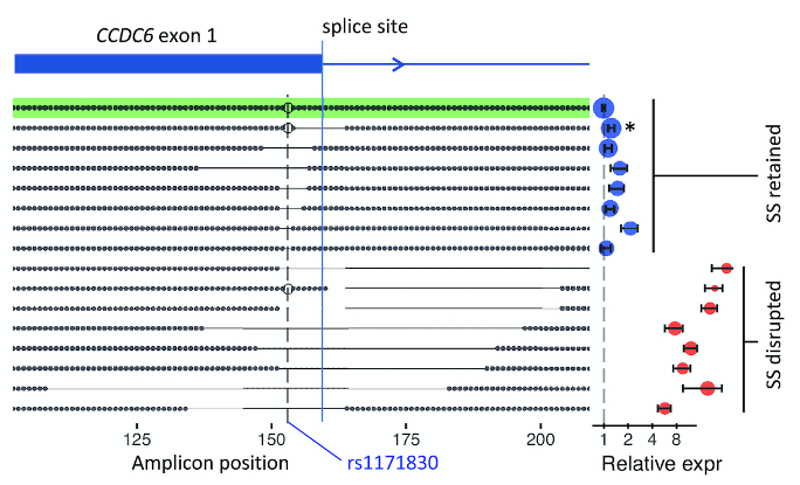
With GenIE it is even possible to edit the genome using a complex mixture of HDR events, and by carrying this out around the splice site we were able to recapitulate the known consensus splicing motif.
There are a few things to keep in mind with GenIE. It is important to use the right cell type to see a relevant effect, and the gene also needs to be transcribed at a high enough level. We think it’s feasible at >= 1 TPM, but this also depends on the editing efficiency of the SNP. As with any CRISPR experiment, not all guides edit efficiently. Importantly, when using GenIE you’re measuring nascent transcripts for intronic SNPs, so you’re only assaying the effect on the gene in which the SNP is located. It’s also important to consider both the number of replicates you do as well as the effect size that you see for each SNP.
Previous methods for finding causal SNPs from GWAS can’t do quite what GenIE does. They either (a) can test a lot of sites across the genome, but can’t tell you about individual SNPs (because they only give you resolution limited to 1000 bp or more if they use KRAB-based repression), or (b) are extremely laborious, since they require isolating multiple clonal cell lines. GenIE fills a sweet spot between the two — you can test the entire “credible set” of potentially causal variants for a GWAS association with single-allele resolution in just a couple of weeks. One of the most important aspects of GenIE is that screening the SNPs of interest occurs at the endogenous location. And, given that the editing is performed in a pool of cells, it is relatively simple to do a pooled differentiation into a relevant cell type where you expect to see a regulatory effect of the SNP on your gene of choice. GenIE could also be applied to primary cells for which sub-cloning or growing is difficult.
We hope that our new method, along with the rgenie analysis package, will be used to identify causal variants in disease-relevant genes, aiding target prioritisation and drug discovery for a large range of human diseases.


