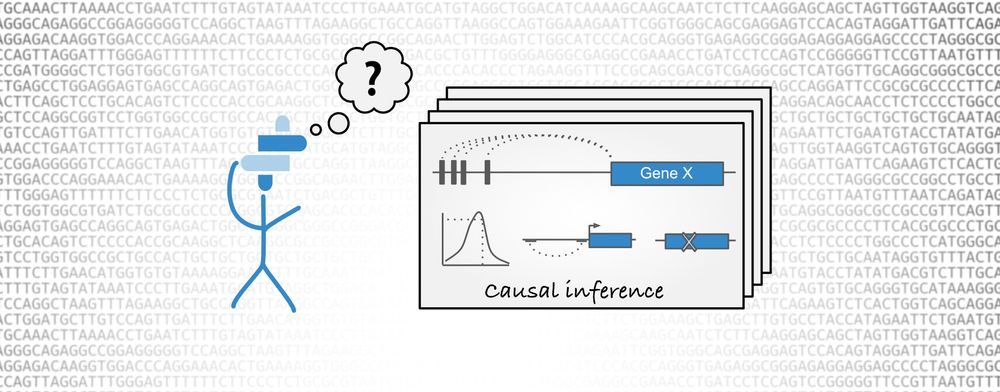
GWAS causal inference in Open Targets informatics tools
Two independent studies indicate drug targets with common disease-associated genetic evidence are twice as likely to lead to an approved drug [1,2].While resources such as GWAS Catalog curate and archive evidence from published literature, pinpointing the causal gene underlying the trait-associated loci remains a major challenge. Moreover, the emergence of large studies such as the UK Biobank has resulted in an unprecedented explosion of knowledge on the number of loci that influence complex traits and diseases.
Open Targets Genetics aims to leverage the increasing amount of publicly available data, in order to identify and prioritise targets with a human genetic basis. The Portal provides a framework to centralise and analyse GWAS data in order to answer two fundamental biological questions: what is the list of likely causal variants at a given trait-associated locus and what is the potential causal gene for a particular trait-associated variant. In the latest release of Open Targets Genetics, we have introduced a new methodology, the Locus-to-gene (L2G), that allows us to systematically identify causal genes at all GWAS associated loci in a phenotype-specific manner. This machine-learning method integrates across the fine mapping credible set, QTL colocalisation and functional genomics data significantly improving our causal gene inference.
Assigning GWAS hits to causal genes
For a given SNP, identifying the causal gene can be quite a complex task. Eric Vallabh Minikel summarised the problem nicely in a recent blog post:
In Mendelian disease, most causal variants are either protein-coding or have other obvious links to a single gene. They are rare, their effect sizes are large, and Mendelian segregation in just a few families can convincingly implicate a gene. GWAS is different. Most hits are common variants of small effect size. Each “hit” is not a single SNP but, rather, a linkage disequilibrium (LD) peak — a whole collection of SNPs that have traveled together with the (often single) true causal SNP on a haplotype block across human generations. And most genome-wide significant SNPs are non-coding, and these variants can sometimes have regulatory effects at a considerable distance
Open Targets Genetics aims to systematically approach gene causal inference from GWAS studies by aggregating across the credible set and enriching the gene assignment with large scale functional genomics datasets. Some of the key features are derived from:
-
Fine mapping — This method helps define a set of variants that are likely to be causal at a given disease-associated locus.
-
Colocalisation — This method tests whether two independent association signals at a locus (e.g. disease-molecular trait) are consistent with having a shared causal variant.
-
Functional genomics — These datasets inform how GWAS-associated SNPs could be mediating their functional effect and the gene they are likely to target. These variant-centric data are integrated with fine mapping information by aggregating across all variants within a given credible set, and weighting by the posterior probability of each.
When it comes to integrating the relevant causal information, the L2G method uses a machine learning algorithm to determine the relative importance of the described features and to derive non-trivial relationships between the data sources. In order to train our gradient boosting model, we required a gold standard set of published GWAS loci for which we have high confidence in the gene mediating the association. We assembled >400 of these examples with different levels of confidence and made this repository GWAS gold standards publicly available. We encourage the genetic community to use this repository for any future benchmarks and to contribute to it as more genes are causally linked to a given disease-associated locus either through robust functional experiments or through other approaches.

Open Targets Genetics Locus-to-gene assignment. Full-size image. Credit: Andrew Hercules.
Boosting target identification and prioritisation
Association studies constitute a fundamental tool to understand the genetic basis of common or complex diseases in a systematic manner. However, the decision-making necessary to start a project or progress through the clinical pipeline often requires multiple lines of evidence. In order to maximise the exploration of orthogonal information, we have populated the Open Targets Platform with all the associations provided in Open Targets Genetics. In the Platform, a visitor can explore genetic evidence, as well as other information from clinical trial studies, animal models, literature mining, transcriptomics or other pathway and Systems Biology analysis. Moreover, the Open Targets Platform will handle overlapping evidence from different studies, in order to increase the confidence of a particular association. A disease ontology expansion algorithm ensures that similar traits are enriched with overlapping signals. Similarly, we are planning to incorporate interaction networks to increase the confidence of our hits, a strategy that has proven successful in previous cases.[3] Altogether, the Platform is the best environment to follow up on some of the leads that genetics might provide.
Up until the 20.02 release of the Open Targets Platform, we received common disease genetic evidence directly from the GWAS Catalog manually curated associations. The most likely causal gene was assigned purely based on distance. With the inclusion of the Open Targets Genetics evidence we have deprecated >40k associations that are no longer considered significant due to the more stringent association p-value threshold (5e-8). Moreover, the replacement of the closest gene assignment by the L2G assignment implies that one SNP might have multiple causal genes with varying scores. Consequently, the number of evidence has grown from 186,237 (19.11 release) to 1,932,925 (20.02 release). Although the increased new evidence causes a dilution of validated target - disease pairs, the L2G method provides a very powerful discrimination score for targets of drugs approved for the same indication. Highly scoring causal genes can outperform the previously reported 2x enrichment in targets for approved drugs.

Benchmark between different GWAS evidence generated from GWAS Catalog and Open Targets Genetics using different variant-to-gene (V2G) methods and thresholds. For comparison purposes, clinical trial data and approved drugs are derived from [2] (Informa Pharmaprojects, Jan 2018). Clinical progression enrichment is calculated as the odds ratio of drugs targeting genes with genetic evidence for the same indication within the universe of gene-indication pairs in clinical trials.
One of the multiple benefits of the Open Targets Genetics feed is the analysis of studies with full summary statistics. For example, the GWAS Catalog curated a study by Tsoi et al.[4] containing 41 loci significantly associated with psoriasis. Since the authors provided full summary statistics, the Open Targets Genetics pipeline inferred an expanded list of 89 independently-associated loci. Interestingly, one of the novel associations (rs77520588) is in close proximity (9,698 base pairs) to the cell adhesion molecule CD2. By visiting the Open Targets Platform, we can not only corroborate that CD2 is transcriptionally up-regulated in psoriasis, but more importantly, that there is an approved antigen drug (Alefacet) indicated for psoriasis targeting CD2.

Association between CD2 and psoriasis seen in the locus plot view in the Open Targets Genetics (left) and the approved drug Alefacet in the drug summary page of the Open Targets Platform (right).
Another advantage of using Open Targets Genetics is the access to the UK Biobank GWAS data. For example, 8 lead variants were associated with an Osteoporosis-related trait in the UK Biobank. Of these, a regulatory variant (rs9594738) is in close proximity to a long non-coding RNA (LINC02341) but there is no additional support. Nevertheless, the third closest gene is the osteoclast differentiation and activation factor (TNFSF11). Supporting eQTL evidence links this variant with changes in gene expression of TNFSF11. Moreover, the Open Targets Platform provides additional evidence that an approved antibody targeting TNFSF11 (Denosumab) has been indicated to treat osteoporosis. Overall, the cumulative evidence points to a more distant gene as the most likely causal gene for this association. This example illustrates the benefits of looking at alternative explanations for causal assignment and the enriched decision-making by using additional data sources.

Association between TNFSF11 and osteoporosis in Open Targets Genetics (left) and the Open Targets Platform (right).
Update on 02/11/2021: Our L2G scoring method was recently published in Nature Genetics. The preprint is also available on BiorXiv.
Mountjoy, E., Schmidt, E.M., Carmona, M. et al. An open approach to systematically prioritize causal variants and genes at all published human GWAS trait-associated loci. Nat Genet (2021). https://doi.org/10.1038/s41588-021-00945-5
References
- Nelson MR, Tipney H, Painter JL, Shen J, Nicoletti P, Shen Y, Floratos A, Sham PC, Li MJ, Wang J, et al.: The support of human genetic evidence for approved drug indications. Nat Genet 2015, 47:856–860.
- King EA, Davis JW, Degner JF: Are drug targets with genetic support twice as likely to be approved? Revised estimates of the impact of genetic support for drug mechanisms on the probability of drug approval. PLoS Genet 2019, 15:e1008489.
- Fang H, ULTRA-DD Consortium, De Wolf H, Knezevic B, Burnham KL, Osgood J, Sanniti A, Lledó Lara A, Kasela S, De Cesco S, et al.: A genetics-led approach defines the drug target landscape of 30 immune-related traits. Nat Genet 2019, 51:1082–1091.
- Tsoi LC, Spain SL, Knight J, Ellinghaus E, Stuart PE, Capon F, Ding J, Li Y, Tejasvi T, Gudjonsson JE, et al.: Identification of 15 new psoriasis susceptibility loci highlights the role of innate immunity. Nat Genet 2012, 44:1341–1348.